四川大学考古科学中心何光林/王萌鸽团队联合多家国内法医学团队揭示中国古今人群父系遗传演化并成功研发超高分辨率法医学体系

原文链接:https://academic.oup.com/mbe/advance-article/doi/10.1093/molbev/msae122/7695223?searchresult=1
在大人群基因组学时代,群体基因组学和人类泛基因组计划正全面记录全球不同人群的遗传图谱,揭示其群体历史,并探究复杂性状及疾病的遗传基础。东亚作为文明的发源地之一,同时是连接大洋洲、西伯利亚和美洲的重要枢纽,其遗传图谱在群体基因组学时代仍然缺乏充分表征。中国拥有丰富的遗传、表型、文化和民族语言多样性,这使其在研究人群复杂历史,包括人类的分化、迁徙和混合,以及遗传与文化的相互作用方面具有独特优势。然而,基于大规模古今基因组资源重建中国古今人群基本演化框架仍需系统解析。
2024年6月17日,《Molecular Biology and Evolution》在线发表了广东省毒品实验技术中心刘超院士团队、南方医科大学朱波峰院长团队,以及四川大学考古科学中心和华西罕见病研究院何光林/王萌鸽团队合作完成题为“Multiple human population movements and cultural dispersal events shaped the landscape of Chinese paternal heritage”的研究论文。研究基于中国万人基因组多样性计划(10K_CPGDP)和炎黄队列(YanHuang cohort)等项目相关的Y染色体基因组资源的初步结果,构建了首个超高分辨率的东亚古今人群整合演化框架。系统解析了古今人群的起源、分歧、瓶颈、扩张及混合事件,并阐述了父系演化进程与语言传播及生业模式转变等社会人类学事件对东亚人群父系遗传结构的影响。此外,研究团队还研发了首个兼顾所有东亚人群父系演化支系的超高分辨率法医学Y染色体检测体系。这一体系有助于加快法医学疑难案件的家系排查和父系生物地理溯源,为犯罪嫌疑人的智能定位和身份识别提供了新技术和新方法。
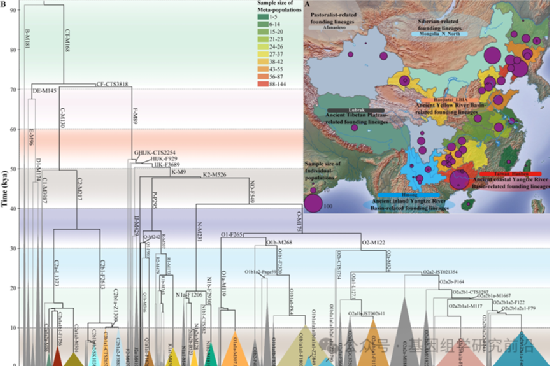
图1 中国人群特异性的高分辨率Y染色体谱系演化框架
目前,众多研究致力于探究中国人群的进化历史及复杂性状和疾病的遗传学基础,以填补人类基因学这一领域的中关于中国人群遗传多样性系统蓝图的知识空白[1,2]。最近的研究利用全基因组SNP芯片,深入分析了汉藏语、蒙古语、通古斯语、突厥语、壮侗语和苗瑶语等不同群体的基因组多样性和群体历史[3-9]。同时,全基因组测序技术的发展推动了多个重要研究项目[10-17],如西湖生物样本库、NyuWa基因组资源、中国代谢解析计划和中国万人基因组多样性计划(10K_CPGDP)。这些项目不仅加深了我们对中国各民族语言群体的基因多样性、群体历史及复杂性状和疾病遗传基础的理解,还为从单系遗传标记和群体规模角度探索其精细遗传结构提供了重要资源。此外,大规模人群基因组项目和古DNA技术的创新,为探索人类演化历史开辟了新途径。然而,古代欧亚人群对中国父系支系格局形成的遗传贡献尚未得到充分研究。
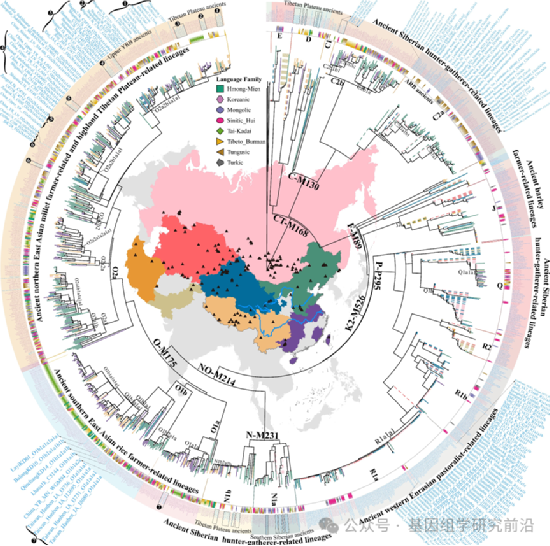
图2 高时空覆盖度的欧亚古今人群整合系统发育关系
Y染色体的非重组区域已成为研究人类父系演化历史的重要工具。随着测序技术和基因组组装技术的进步,以及读长比对、变异分型和基准测试等计算方法的发展,我们获得了Y染色体的完整序列,大大丰富了对其遗传变异的认识。这些成果促进了稳定系统发育树的构建。过去二十年中,基于靶向Y-SNPs的研究通过追踪父系支系,为研究人类的起源、迁徙和混合提供了关键数据。采用先进的下一代测序技术和计算生物学方法对Y染色体进行全序列重测序,已改变了Y染色体研究的范式。然而,中国人群的大规模Y染色体基因组数据库仍然有限,这凸显了建立更全面数据库的必要性,以探索中国精细父系遗传结构及其历史影响因素。
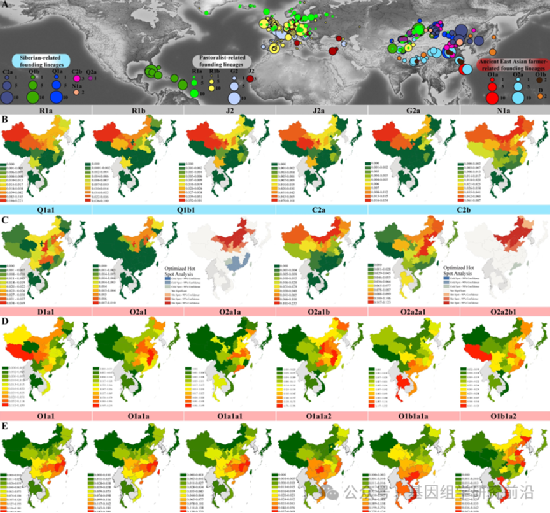
图3 欧亚Y染色体古基因组资源的分布及现代族群中关键奠基者父系的频率图谱
近年来,中国人群基因组资源的快速增长的同时,也反应了我们在理解其父系遗传多样性方面的不足。与汉族及全球其他人群的研究相比,民族语言多样性族群的精细演化历史领域的进展明显滞后。为弥补这一差距,我们启动了10K_CPGDP项目,采用人类学田野调查采样策略,并引入了炎黄队列(YHC)基因组资源。YHC项目不仅包含新生成的Y染色体序列,还整合了10K_CPGDP中的Y染色体数据。其目标是建立高质量的特定人群Y染色体数据库,精细描绘代表性不足群体的父系历史,构建高分辨率、带时间轴的系统发育树,并开发高分辨率的东亚人群特异性单倍型参考面板和法医基因组学体系研发、数据库构建和算法开发。基于大规模人群基因组资源的理论研究和转化运用将用于医学基因组学、法医学精准祖源推断和大规模远距离家系重建等应用。
本项目开发了“YHSeqY3000”超高分辨率Y染色体检测系统,这是目前最精确的Y特异性靶向重新测序法医学检测系统,基于YHC的全基因组数据和芯片数据设计。研究团队利用YHSeqY3000对919名中国少数民族男性进行基因分型,并通过Y染色体全基因组测序验证其结果。通过整合已发表的古今基因组数据,我们建立了一个包含15,563名现代和古代欧亚个体的Y染色体数据库,并构建了首个完整且高分辨率的古今人群综合系统发育树,包括中国不同时期的古基因组序列。这一系统发育树有助于估计主要父系支系的分歧、瓶颈事件、扩张和混合的发生时间,追踪中国不同父系支系的起源,揭示历史上的迁徙、混合和生计策略变化对中国各人群父系遗传结构的影响。
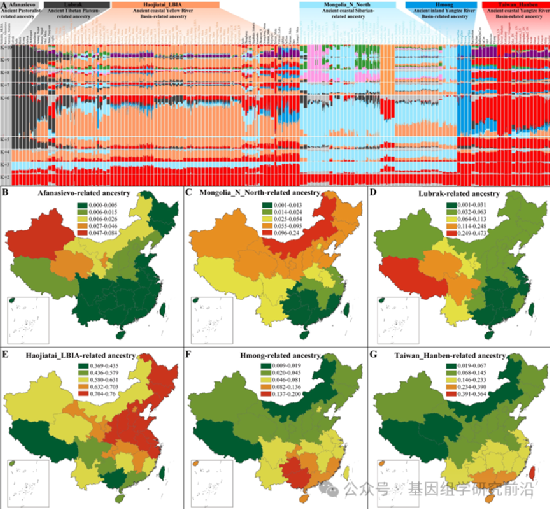
图4 东亚古今族群遗传混合模型及关键常染色体模拟祖源相关遗传成分的分布模式
整体而言,本研究构建了一个综合的Y染色体古今人群基因组数据库,涵盖了15,563名现代和古代欧亚个体,以探索中国的父系基因组多样性。通过构建高分辨率且具有时间标记的系统发育树,我们揭示了新石器时代早期和中期的多次父系支系分化和大规模扩张事件。研究结果阐明了四次主要的古代群体迁徙,这些迁徙均与生业技术的创新紧密相关,对中国现代父系遗传格局的形成产生了深远影响。首先,早期东亚人和黄河流域的粟作农民携带O2/D亚支系的扩张,显著影响了汉藏语人群父系支系格局,促进了人群在青藏高原的永久定居。其次,携带O1和某些O2亚支系的长江流域稻作农民的扩散,重塑了南方汉族以及壮侗语、南岛语、苗瑶语和南亚语人群的父系遗传多样性。第三,起源于新石器时代西伯利亚的Q/C等父系支系,在蒙古高原和黑龙江流域的狩猎采集者中兴起和扩散,对中国北方人群的基因库产生了显著影响。第四,起源于西欧亚的J/G/R等父系支系,最初由与颜那亚文化有关的草原游牧人群传播,主要存在于中国西北地区。总体而言,本研究提供了全面的遗传学证据,揭示了不同文化背景下古代欧亚人群的互动对现代中国人群父系遗传多样性格局的显著影响。基本框架的解析,为研究团队下一步精细解析人类Y染色体序列隐藏的基因组医学和群体演化奥秘提供方向。
四川大学考古科学中心/四川大学华西罕见病研究院王萌鸽博士、黄雨果博士后和何光林副研究员为共同第一作者,广东省毒品实验技术中心(国家毒品实验室广东分中心)的刘超院士、南方医科大学朱波峰院长、四川大学考古科学中心/四川大学华西罕见病研究院王萌鸽博士和何光林副研究员为共同通讯作者。相关工作得到国家自然科学基金、四川大学考古科学中心开放课题等项目的资助。
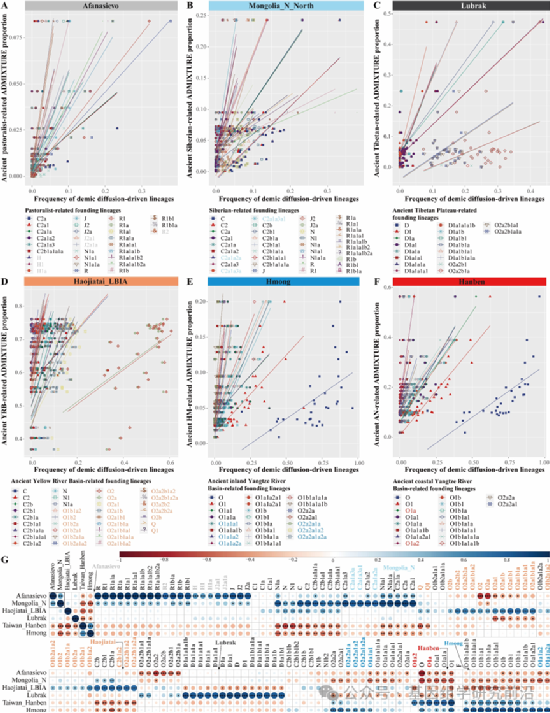
图5 东亚关键奠基者父系支系的多因素相关性分析
延伸阅读相关参考文献:
[1] Yang MA, Fan XC, Sun B, Chen CY, Lang JF, Ko YC, et al. Ancient DNA indicates human population shifts and admixture in northern and southern China. Science 2020;369:282-+.
[2] Wang CC, Yeh HY, Popov AN, Zhang HQ, Matsumura H, Sirak K, et al. Genomic insights into the formation of human populations in East Asia. Nature 2021;591:413-+.
[3] Sun Q, Wang M, Lu T, Duan S, Liu Y, Chen J, et al. Differentiated adaptative genetic architecture and language-related demographical history in South China inferred from 619 genomes from 56 populations. BMC Biol 2024;22:55.
[4] He GL, Wang PX, Chen J, Liu Y, Sun YT, Hu R, et al. Differentiated genomic footprints suggest isolation and long-distance migration of Hmong-Mien populations. Bmc Biology 2024;22:18.
[5] He G, Wang Z, Guo J, Wang M, Zou X, Tang R, et al. Inferring the population history of Tai-Kadai-speaking people and southernmost Han Chinese on Hainan Island by genome-wide array genotyping. Eur J Hum Genet 2020;28:1111-23.
[6] He G, Wang M, Luo L, Sun Q, Yuan H, Lv H, et al. Population genomics of Central Asian peoples unveil ancient Trans-Eurasian genetic admixture and cultural exchanges. hLife 2024.
[7] Li X, Wang M, Su H, Duan S, Sun Y, Chen H, et al. Evolutionary history and biological adaptation of Han Chinese people on the Mongolian Plateau. hLife 2024.
[8] Sun Y, Wang M, Sun Q, Liu Y, Duan S, Wang Z, et al. Distinguished biological adaptation architecture aggravated population differentiation of Tibeto-Burman-speaking people. J Genet Genomics 2024;51:517-30.
[9] Wang MG, He GL, Zou X, Chen PY, Wang Z, Tang RK, et al. Reconstructing the genetic admixture history of Tai‐Kadai and Sinitic people: Insights from genome‐wide SNP data from South China. Journal of Systematics and Evolution 2022;61:157-78.
[10] Cao YN, Li L, Xu M, Feng ZM, Sun XH, Lu JL, et al. The ChinaMAP analytics of deep whole genome sequences in 10,588 individuals. Cell Research 2020;30:717-31.
[11] Feng Y-CA, Chen C-Y, Chen T-T, Kuo P-H, Hsu Y-H, Yang H-I, et al. Taiwan Biobank: A rich biomedical research database of the Taiwanese population. Cell Genomics 2022;2:100197.
[12] Wang C, Dai J, Qin N, Fan J, Ma H, Chen C, et al. Analyses of rare predisposing variants of lung cancer in 6,004 whole genomes in Chinese. Cancer Cell 2022;40:1223-39 e6.
[13] Cheng S, Xu Z, Bian SZ, Chen X, Shi YF, Li YR, et al. The STROMICS genome study: deep whole-genome sequencing and analysis of 10K Chinese patients with ischemic stroke reveal complex genetic and phenotypic interplay. Cell Discovery 2023;9:75.
[14] Walters RG, Millwood IY, Lin K, Valle DS, McDonnell P, Hacker A, et al. Genotyping and population characteristics of the China Kadoorie Biobank. Cell Genomics 2023;3:100361.
[15] Huang SJ, Liu SY, Huang MX, He JR, Wang CR, Wang TY, et al. The Born in Guangzhou Cohort Study enables generational genetic discoveries. Nature 2024;626:565-+.
[16] Jiang T, Guo H, Liu Y, Li G, Cui Z, Cui X, et al. A comprehensive genetic variant reference for the Chinese population. Science Bulletin 2024.
[17] Zhang P, Luo H, Li Y, Wang Y, Wang J, Zheng Y, et al. NyuWa Genome resource: A deep whole-genome sequencing-based variation profile and reference panel for the Chinese population. Cell Rep 2021;37:110017.