Center for Archaeological Science, Sichuan University published a review on new paradigms and research progress of mitochondrial genome research in the context of large-scale population genome sequencing in SCLS.

On September 17, 2024, the team of Professor Yuan Huijun/Associate Researcher He Guanglin from the Center for Archaeological Science, Sichuan University and the Institute of Rare Diseases, West China Hospital, Sichuan University, jointly with the research team of Academician Liu Chao from the Guangdong Drug Experiment Technology Center (Guangdong Branch of the National Drug Laboratory), and the team of Professor Tang Renkuan from the School of Basic Medical Sciences, Chongqing Medical University, published a review paper entitled "Sequencing and characterizing human mitochondrial genomes in the biobank-based genomic research paradigm" in SCIENCE CHINA Life Sciences. This study systematically reviewed the latest advances in human mitochondrial DNA (mtDNA) research in the new paradigm of human genomics research supported by large-scale biobanks, including mtDNA heteroplasmy, maternal inheritance patterns, paternal inheritance controversies, nuclear mitochondrial DNA segments (NUMTs), mitochondrial DNA regulation mechanisms, new mechanisms of mitochondrial diseases, and the construction of mitochondrial databases. The study emphasized the critical roles of next-generation sequencing (NGS) technology and large-scale mtDNA data resources in analyzing mtDNA variations, exploring NUMTs functions, and understanding mtDNA in human evolution, and looked forward to the importance of interdisciplinary collaboration in promoting research in the field of human genetics, while pointing out the potential of third-generation sequencing technology (TGS) and artificial intelligence in genome research.

This review focuses on three exciting areas of mtDNA research: the atlas of mitochondrial heteroplasmy, the distribution and impact of NUMTs, and the genetic diversity and maternal evolutionary history of mtDNA. With the development of next-generation sequencing technology (NGS), the advancement of the Human Genome Project, and the establishment of large-sample bio-databases, our understanding of mtDNA-related scientific issues is constantly being revised and refined. For example, the widespread existence and inheritance patterns of mitochondrial heteroplasmy in normal humans, the distribution and generation processes of NUMTs, and the key roles of mtDNA variant sites in tracing early human migration and understanding regional human biological adaptation. Finally, this review looks forward to how multidisciplinary collaboration will deepen our understanding of the role of mtDNA in precision medicine, genetics, and the study of human evolutionary history, providing new perspectives and methods for solving long-standing problems in clinical medicine and anthropology, in the context of the rapid development of TGS and the massive production of genomic data.
Mitochondria are the energy factories of cells, and mtDNA encodes key proteins of the oxidative phosphorylation system, which plays an important role in regulating oxidative respiration and life activities. There are multiple mtDNA copy numbers in a cell, and these mtDNAs may be of different types, which is called mitochondrial heteroplasmy. Variant mtDNA can lead to mitochondrial dysfunction, which in turn causes a variety of metabolic and degenerative diseases. Growing evidence suggests that mitochondrial heteroplasmy is widespread and has complex effects on human physiological function. In addition, there are various interactions between mtDNA and nuclear DNA (nDNA), and NUMTs are direct evidence of genetic material transfer at the cellular level. In addition, the characteristics of maternal inheritance and high mutation rate of mtDNA make it a powerful tool for studying human origin and migration patterns, which helps to answer key questions in human migration history.
MtDNA is essential for understanding cellular energy metabolism, numerous human diseases, and evolutionary dynamics. Unlike nDNA, mtDNA is maternally inherited, has a higher mutation rate, and undertakes unique functions, providing us with a unique perspective on basic biological processes. Despite its simple structure, mtDNA encodes fourteen key proteins located in the oxidative phosphorylation system (OXPHOS), which are essential for energy conversion in human metabolism (Figure 1a-b). Mutations in these coding genes can lead to mitochondrial dysfunction, thereby reducing ATP production and increasing reactive oxygen species. These changes are characteristic of various metabolic and degenerative diseases. MtDNA has broad importance in disease research, involving both mitochondrial diseases caused by direct mtDNA mutations and complex diseases associated with indirect mitochondrial dysfunction. Mitochondrial diseases (MDs) exhibit diverse symptoms and inheritance patterns, emphasizing the role of mtDNA in cellular health. The existence of heteroplasmy increases the complexity of disease phenotypes and inheritance, posing challenges for genetic and clinical research. On the other hand, mtDNA is also a powerful evolutionary tool for studying human origin and migration patterns because it is maternally inherited and has no recombination (Figure 1d). Its high mutation rate has been used as a molecular clock (Figure 1c), playing a huge role in reconstructing phylogenetic relationships and human population history. In addition, the emergence of large-scale bio-databases and advances in sequencing technology have facilitated the study of mtDNA. The high-quality biosamples in bio-databases provide the necessary genetic material for comprehensive mtDNA research. At the same time, NGS and TGS technologies enable rapid, high-resolution sequencing and analysis of mtDNA, allowing for more precise characterization of its variations in different populations and diseases. This synergy of biobanks and advanced sequencing technologies marks a paradigm shift in genomic research, enhancing our understanding of the role of mtDNA in health, disease, and human history (Figure 2).
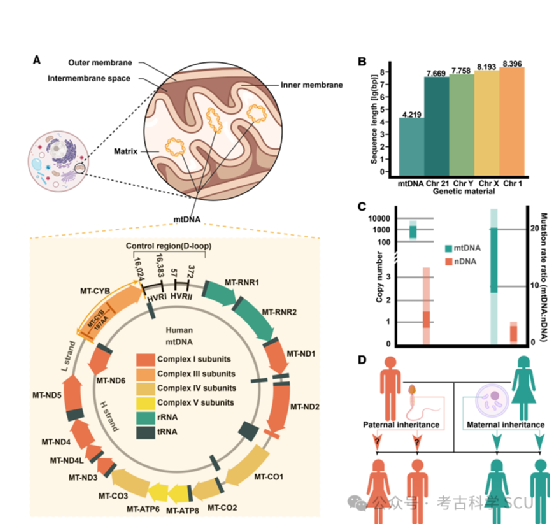
Figure 1 Structure and genetic characteristics of mtDNA.
MtDNA Heteroplasmy Atlas
Mitochondrial DNA heteroplasmy can be divided into length heteroplasmy and sequence heteroplasmy (Figure 3a-b). Length heteroplasmy is usually caused by insertion/deletion (InDels) or replication, which changes the length of the mtDNA molecule; while sequence heteroplasmy involves changes in the nucleotide sequence of the same individual, mainly single nucleotide variations (SNVs). These variations may stem from internal factors, such as DNA replication mismatches, or external factors, such as ultraviolet light, radioactive radiation, chemicals, and certain viruses. Studies have shown that there is a correlation between age and heteroplasmy, and animal studies have also found gender bias in heteroplasmy. These findings have sparked discussions about which heteroplasmies are genetically acquired, which are related to cell proliferation and differentiation, and the extent to which heteroplasmy is regulated by nuclear DNA. Previous studies have determined that severe and pathogenic mtDNA mutations in germ cells are mainly maternally inherited, although they can also be de novo. However, the recurrence risk of de novo mutations is low, which emphasizes the critical role of prenatal counseling.
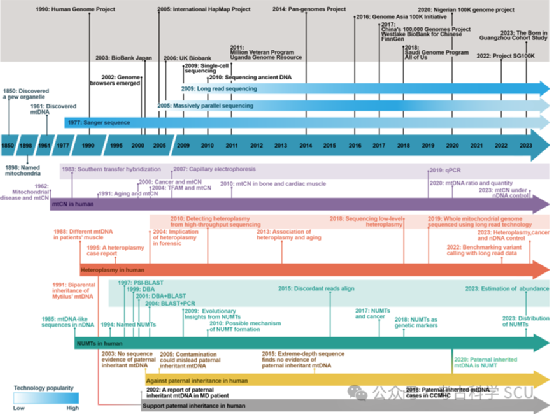
Figure 2 Timeline of mtDNA research and technological advances
Recent studies have filled in the gaps in our understanding of mtDNA heteroplasmy. Gupta et al. analyzed the heteroplasmy of approximately 300,000 people in the UK Biobank and the US All of Us datasets, and found that heteroplasmic SNVs accumulate with age and tend to be of somatic origin, while heteroplasmic InDels are maternally inherited. In addition, genome-wide association studies have revealed that multiple nuclear genes are involved in regulating heteroplasmy levels. MtDNA heteroplasmy is also critical in the pathogenesis of MDs, and its pathogenesis is significantly different from that of nDNA variations. Although many mtDNA mutation sites are associated with MDs, the threshold effect still needs to be considered during diagnosis. This effect refers to the fact that heteroplasmic mtDNA mutations need to reach a critical level to disrupt mitochondrial function and trigger disease symptoms. This critical level significantly affects the onset and severity of mitochondrial diseases (MDs) (Figure 3b). The threshold is not uniform between individuals and is affected by factors such as the type of mutation, its location, and the energy requirements of different tissues. Therefore, some individuals with high levels of heteroplasmic mutations may be asymptomatic, while others with lower levels show symptoms. Understanding the threshold effect is essential for accurately diagnosing MD and elucidating the relationship between genetic variations and mitochondrial function.
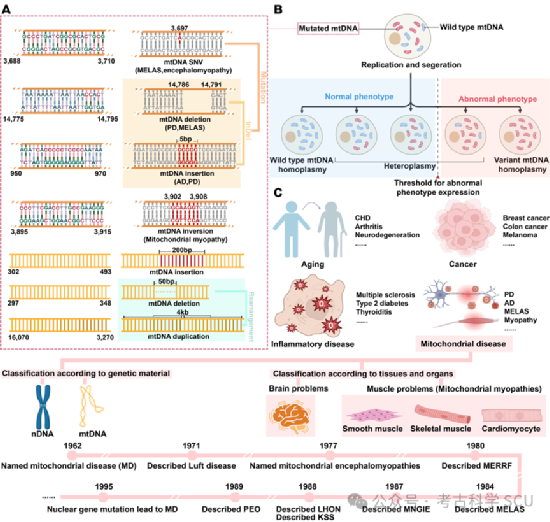
Figure 3 Types of mtDNA variations and classification of mitochondrial diseases
Studies have linked heteroplasmy levels to a variety of diseases, including type 2 diabetes, stroke, and hypertension (Figure 3c). In many cancers, heteroplasmic variations accumulate. The clinical manifestations of heteroplasmy may mask the genetic basis, complicating the diagnosis of MDs and delaying treatment. Therefore, it is necessary to understand the distribution and inheritance patterns of heteroplasmy at the population level. Heteroplasmy levels can be defined relatively (the ratio of mutant mtDNA to total mtDNA) or absolutely (the number of mutant mtDNAs), and most studies focus on the former. It was previously thought that non-synonymous heteroplasmic mutations may exist in healthy individuals, but pathogenic heteroplasmy should not exist. However, this view has changed. A key study showed that 1/192 individuals carried pathogenic variations without obvious clinical symptoms. Single-cell sequencing and large-scale population genomic data have revealed dynamic fluctuations in heteroplasmy within individual cells. These studies indicate that heteroplasmy can be detected in healthy individuals, so MDs have a complex connection with heteroplasmy.
This shift in perceptual paradigm is largely attributed to the rapid development of NGS technology. The earliest Sanger sequencing methods were limited in their application to complete mitochondrial genome sequencing due to their low throughput and high cost. Therefore, early mtDNA sequencing mainly focused on hypervariable regions I (HVR I: 16024-16383) and II (HVR II: 57-372), ignoring regions with high GC content and secondary structure domains. This reduced sequencing resolution, hindering the accurate detection of genetic differences between individuals and the exploration of heteroplasmy within the same individual. Although NGS is prone to false positives or false negatives, its high-throughput characteristics and expanded reading range increase data density, allowing for more detailed sequencing of the entire mitochondrial genome and facilitating the detection of low-level heteroplasmy. Guo et al. identified the inheritance patterns and bottleneck effects of variations in mother-child pairs. Subsequent studies have adopted various quality control methods, gradually reducing the detection limit of heteroplasmy levels to below 1% or even 0.5%. One area worth emphasizing is the study of the relationship between low-frequency heteroplasmic mutations and aging and reproductive disorders. Although the relationship between heteroplasmic mutations and aging has been partially elucidated, there are still many complex mechanisms that need to be understood. In summary, in-depth research on mtDNA heteroplasmy not only helps us understand human genetic diversity, but also promotes the development of precision medicine and personalized medicine, and promotes early disease diagnosis and prevention. Although more experimental verification is needed, current research has provided new perspectives for our scientific exploration.
Patterns and Functions of NUMTs
In the human nucleus, there is a special class of DNA sequences called NUMTs. They are highly similar to mtDNA sequences but exist in the nucleus. The discovery of these sequences gives us a deeper understanding of the genetic exchange within cells. The origin of NUMTs can be traced back to 1967, when mtDNA-like sequences in the nuclear genome were first discovered through hybridization experiments. It was not until 1994 that Lopez et al. officially named these fragments NUMTs through sequencing and comparison. A complex and fascinating question followed: How is mtDNA integrated into the nuclear genome? Mitochondria may leak mtDNA into the cytoplasm during morphological changes such as division, fusion, expansion, contraction, fragmentation, and autophagy, which in turn leads to the transfer of mtDNA fragments to the nuclear genome (Figure 4a-b). Some studies have shown that the interaction between mtDNA and nDNA may be related to the repair of nDNA double-strand breaks (DSBs). It is now generally believed that the formation of NUMTs is closely related to the non-homologous end joining (NHEJ) pathway (Figure 4b).
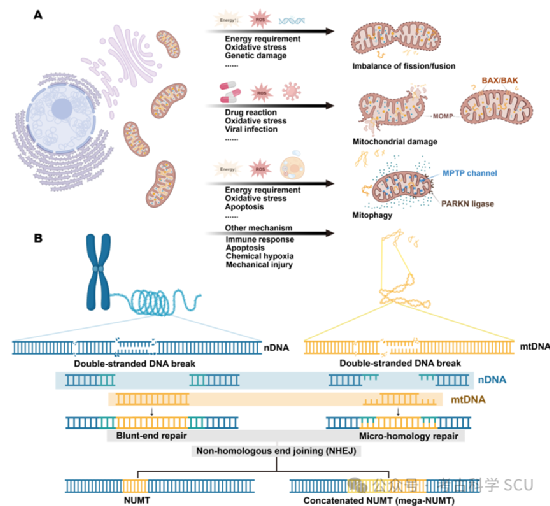
Figure 4 Formation mechanism of NUMTs
Accurate identification of NUMTs is crucial because misinterpreting them as mtDNA heteroplasmy can lead to misdiagnosis of MDs and unreliable interpretation of heteroplasmic variations. Methods for distinguishing NUMTs from mtDNA heteroplasmy can be divided into wet experimental methods and computational analysis methods, each with its own advantages and disadvantages. Wet experimental methods, such as mitochondrial isolation and PCR primer design targeting NUMTs, provide intuitive and accurate results. However, these techniques are expensive and time-consuming, and may not capture the full diversity of NUMTs. Therefore, computational methods are more commonly used. Early methods used the Basic Local Alignment Search Tool (BLAST) to detect and exclude nDNA sequences similar to mtDNA sequences. Researchers later used the distribution and frequency of k-mers to identify potential NUMTs. Current research often combines a variety of bioinformatics tools to create personalized, efficient, and flexible workflows. However, computational methods are prone to false positives and false negatives, so it is crucial to use statistical measures such as mtCN, variant allele frequency, and quality scores to validate the results.
NUMTs are ubiquitous in eukaryotic genomes, and the fragment length and number vary between different species. Some NUMTs are derived from the entire mitochondrial genome, indicating complex interactions between mitochondrial DNA and nuclear DNA. Initially, NUMTs were considered relics of ancient endosymbiotic processes about 2 billion years ago. However, the latest research shows that the emergence of NUMTs has run through the entire human evolution process. This discovery indicates an important intracellular gene transfer pathway and enhances our understanding of the endosymbiotic hypothesis. In addition, most NUMTs are non-functional and are mainly located in genomic repetitive regions, regulatory elements, short interspersed nuclear elements (SINEs), simple repeat sequences, and introns. NUMTs are rarely found in coding regions or within 500 bp of gene transcription start sites. This distribution may stem from the site specificity of DSB repair or significant selective pressure on coding regions. Studies have also revealed a negative correlation between gene intolerance scores and NUMTs frequency, suggesting that NUMTs near genes may be subject to strong evolutionary selection pressure to maintain gene structure and functional stability.
Although NUMTs are mainly non-functional, understanding their distribution and dynamic changes in different populations is of great significance for preventing misdiagnosis of MDs and understanding diseases and cancer. The fragment length and number of NUMTs vary in different populations, which may reflect the evolutionary history under different genetic backgrounds. Some studies have shown that certain NUMTs are closely related to tumorigenesis, which may indicate that extreme conditions, such as cancer cell proliferation, can trigger NUMT formation or genetic rearrangement. For example, the FUS-DDIT3 gene, which is produced by complex rearrangements involving NUMT insertion, is present in 90% of myxoid liposarcomas, highlighting the carcinogenic effect of NUMTs. In-depth research on NUMTs is essential for revealing key pathways of intracellular gene transfer, providing new insights into the endosymbiotic theory, and understanding the genetic variations and adaptive evolution of humans and their ancestors.
Unexpected Discovery of Paternal Inheritance Patterns
Traditionally, mtDNA is considered to be strictly maternally inherited, meaning that individuals inherit mtDNA from their mothers. However, some studies have proposed evidence that mtDNA may be paternally inherited. In 1991, Gyllensten et al. proposed the view that mtDNA may be inherited through paternal inheritance based on experiments on inbred mouse strains. Subsequently, similar findings have been observed in fruit flies, bees, cicadas, chickens, sheep, and cloned cattle, which has prompted scientists to further study the inheritance patterns of human mtDNA. Luo et al. sequenced mtDNA in multiple generations of multiple families, discovered high levels of heteroplasmy, and proposed the possibility of biparental transmission of mtDNA, and the transmission pattern is similar to autosomal dominant inheritance. This discovery provides evidence for paternal inheritance of human mtDNA, but also brings potential complexity to understanding MDs. However, many studies have failed to replicate these findings, leading to skepticism in the field. Pyle et al. used triplicate mtDNA sequence data with approximately 1.2 million-fold coverage in a study and found no evidence of paternal transmission. With the progress of NUMTs research in the NGS era, a consensus has gradually been reached on the debate about paternal inheritance of human mtDNA. As part of nDNA, NUMTs follow an autosomal inheritance pattern, which complicated early sequencing results and led to erroneous conclusions about paternal inheritance. Short reads may lead to assembly difficulties, reduce mapping accuracy, and hinder the identification of NUMTs. Therefore, optimized NUMTs detection processes and high-coverage WGS data, especially data from PacBio SMRT or Oxford Nanopore technologies, are essential for distinguishing NUMTs from true mtDNA variations.
Maternal Evolutionary History and Genetic Diversity of mtDNA
Since the Neolithic agricultural innovations of human society, subsistence patterns and settlement patterns such as patrilocal or matrilocal residence have significantly affected the paternal and maternal fine-grained genetic backgrounds and formation patterns of human groups. Evolutionary history is crucial for understanding the origins, migrations, admixtures, and genetic diversity patterns of different human languages and ethnic groups. mtDNA, as an ancient and maternally inherited marker, has accumulated enough mutations over time to be used to distinguish genetically distinct groups. The location and number of these mutations can determine the differences between groups, thereby elucidating the phylogenetic relationships between groups. In 1987, Cann et al. sequenced and analyzed 370 mtDNA restriction sites from 147 individuals worldwide, revealing that the most recent common maternal ancestor of modern humans, the "Mitochondrial Eve", originated from a small group in Africa. Although this study provided a broad overview of the genetic composition of modern humans on different continents, it lacked a detailed understanding of the genetic structure of populations in different geographic regions. HVRs are rich in mutations, but only account for a small portion of the mitochondrial genome, making local sequencing insufficient to reconstruct detailed phylogenetic relationships and explore the fine-scale genetic structure between populations. The development of NGS technology has solved these challenges, enabling us to discover and understand genetic diversity more deeply. For example, early targeted sequencing data indicated that a single out-of-Africa event allowed humans to settle in Australia and Eurasia, because the most recent common ancestor times of the three major macrohaplogroups (M, N, and R) in these regions were similar. However, whole-genome sequencing revealed that the divergence times between Australian Aborigines and Eurasian populations were significantly different, approximately 70,000 years ago and 30,000 years ago, respectively, refuting the earlier hypothesis.
Complete mtDNA sequencing also highlighted the process of modern humans rapidly migrating from Africa to Asia along the "Southern Route". Subsequent studies refined this route, including expansions from southern China to the Himalayas and from Southeast Asia along river systems into East Asia. In addition, whole-genome sequencing of 19,568 mtDNA sequences from northern Pakistan and surrounding populations elucidated the genetic impact of Yamnaya-related steppe populations on South Asian populations, as well as the deep connections between the maternal ancestors of Northeast Asia, Japan, and Native American populations. Many studies have described human migration history in detail and elucidated the genetic diversity formed through complex population admixture processes. For example, whole-mtDNA sequences of Tibetan populations in western China confirmed that their composition includes Paleolithic maternal populations and Neolithic millet-farming populations from the Yellow River basin, refining previous archaeological views and empirically demonstrating the common origin of the Sino-Tibetan from the perspective of maternal genetic markers. Similarly, indigenous populations in southern China and Southeast Asia have both ancient genetic components and newly formed genetic components since the Holocene. Overall, whole-genome sequencing has significant advantages in answering human history.
Human Diseases and Phenotypic Variations from an Evolutionary Perspective
From an evolutionary perspective, human diseases and phenotypic variations are the result of constraints, trade-offs, mismatches, and conflicts triggered by natural selection in the process of complex biological systems adapting to changing environments. This article aims to illustrate this view by studying mitochondrial DNA mutations. When we analyze mutations that define different haplogroups, we raise a fascinating question: Do these mutations lead to functional differences and further affect an individual's adaptation or disease in a specific environment? How do these factors affect the current geographic distribution of populations? Some studies have attempted to answer this question. Studies on non-synonymous and synonymous mutations in the mtDNA coding region have shown that ATP6, cytochrome b, and cytochrome c oxidase I (genes that play important roles in OXPHOS) exhibit significant functional differences in populations at different latitudes, suggesting that there may be a link between mutations and the distribution of genetically diverse populations. Specifically, haplogroups M and N dominate in Asia, and two amino acid variations (ND3 and ATP6) of the northern haplogroup N alter the mitochondrial membrane potential and Ca2+ levels, affecting coupling efficiency and increasing heat production, which may provide an adaptive advantage in cold environments. Studies have shown that haplogroups A, C, and D prevalent in Siberia have higher basal metabolic rates than haplogroups B and F. MDs also show unique distribution patterns. For example, Leber's hereditary optic neuropathy (LHON) associated with mutations at positions 11778 or 14484 has a higher risk of disease in haplogroup J, which is widely distributed in Europe, than in other haplogroups. In addition, mutations at positions 16126 and 73 are more likely to cause age-related macular degeneration and retinal pigment problems in the genetic background of haplogroups U and J. Although the risk of myocardial infarction is higher in East Asian populations associated with haplogroup N9a, it is also associated with a reduced risk of diabetes. In the Han Chinese population, haplogroup M7b1a1 is associated with lower BMI, waist circumference, and waist-to-hip ratio, indicating a reduced risk of obesity. However, it also leads to increased levels of total cholesterol and low-density lipoprotein cholesterol.
Sequencing Advances, Genome Databases, and Analysis Techniques
Accurately identifying individual heteroplasmy levels, elucidating the formation mechanisms of NUMTs, reshaping evolutionary history, and advancing personalized medicine and precision treatment all depend on the collection, storage, and analysis of biological data. This poses challenges to sequencing technology, databases, and computational methods. The paradigm shift brought about by NGS has significantly enhanced our understanding of the above scientific issues, but NGS still has limitations. Distinguishing true heteroplasmic variations from false positives caused by NUMTs remains challenging. For example, PCR amplification of DNA samples may introduce biases, affecting the sensitivity of low-level heteroplasmy. In addition, the short reads generated by NGS are not conducive to sequencing the entire mitochondrial genome at one time, nor are they conducive to identifying variations in long NUMTs or complex genomic regions. This complicates accurate mapping in the presence of many NUMTs and may lead to potential errors. In contrast, TGS, despite its limitations, provides longer reads, eliminates the need for PCR amplification, and improves the accuracy of detecting low-level heteroplasmy, structural variations, and NUMTs. To fully utilize TGS technology, two urgent needs must be addressed: establishing high-quality genomic bio-databases that include different languages and ethnic groups and comprehensive disease spectra, and developing optimized algorithms to quickly and accurately identify heteroplasmy and mtDNA variations, as well as NUMTs.
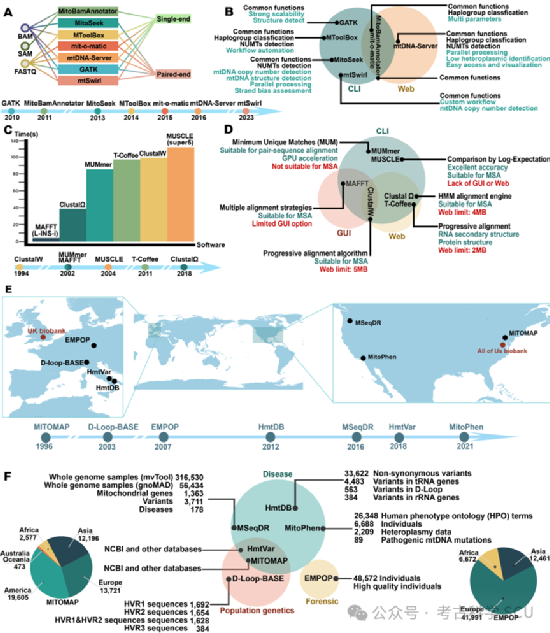
Figure 5 Characteristics of mtDNA databases and analysis tools
In the genomic era, processing and analyzing the massive amounts of data generated has become an increasingly serious issue (Figure 5e-f). Analyzing such a large amount of data requires significant computational resources and complex bioinformatics tools. A key challenge is aligning sequences of variable lengths, and many studies have developed multiple tools and processes (Figure 5c-d). In general, all software programs perform base quality assessment, read alignment, and heteroplasmy level calculation. We summarize some of the currently popular tools and point out their limitations. For example, MitoBamAnnotator only accepts single-ended sequence input, which may miss heteroplasmy data on other DNA strands (Figure 5a-b). Although mit-o-matic has Web and command-line (CLI) interfaces to meet diverse analysis needs, it is affected by too many parameter options, often leading to suboptimal and unreliable results. In addition, using an integrated process of various software through CLI requires manual configuration of a comprehensive runtime environment, which poses significant challenges and reduces the efficiency of data transmission, analysis, and interpretation.
Although current research has made significant progress in building high-quality bio-databases and developing corresponding software, the field still faces many challenges. These challenges include the interpretation and management of noise or information loss in large-scale genomic data due to technical limitations, and the difficulty of combining genetic information with complex real-world environments. Artificial intelligence (AI) provides promising solutions to these challenges. As a branch of AI, machine learning enables computers to improve the efficiency and accuracy of data processing by learning from data and algorithms without explicit programming. This includes techniques for automatically identifying patterns in data distribution, which can be used for prediction or decision-making. Deep learning techniques have shown great potential in identifying patterns and signals in complex data. For example, Min et al. used deep learning methods to analyze human genomic data and successfully predicted the genetic risk of various complex diseases. Angermueller et al. analyzed gene expression data through deep learning models and accurately identified expression patterns associated with specific diseases. Poplin et al. developed a high-precision variant detection tool using deep learning methods to identify SNVs and InDels from high-throughput sequencing data, providing an effective means for genetic research and variant interpretation. In addition, integrated learning methods can improve prediction accuracy and robustness by combining multiple models. In addition, improving the interpretability of algorithms is another key direction of research. Developing models that can process heterogeneous data and consider the interaction between genetic information and the real world is of great significance for solving problems in complex biological systems. In the future, AI will play an increasingly important role in interpreting genetic information, advancing personalized medicine, and understanding the genetic basis of human diseases, and achieving these goals requires interdisciplinary collaboration between computational science and genetics.
Conclusions and Outlook
This review re-examines the latest advances in mtDNA research in medical science and evolutionary genomics facilitated by NGS technology. We highlight the impact of heteroplasmy on mitochondrial function and its association with MDs, as well as the role of NUMTs in challenging traditional mtDNA inheritance concepts and enhancing our understanding of the endosymbiotic hypothesis. In addition, we emphasize that large-scale population mtDNA research has revealed extensive human migration paths and adaptations to different environments, shaping our current maternal genetic structure. This article also examines the characteristics of contemporary computational tools and emphasizes that these tools have improved multiple sequence alignment, improved the accuracy of variation identification, and deepened our understanding of the relationship between structure and function. These advances have refined our knowledge of the role played by mtDNA variations in biological processes. Finally, we emphasize the importance of establishing a comprehensive, high-quality, and accessible mtDNA database that includes common and rare genetic variations.
TGS technology, with its long reads, single-molecule sequencing, and high-resolution structural variation detection capabilities, is critical to achieving our research goals. This method helps to detect extremely low levels of heteroplasmy and enhances the identification of NUMTs distributions, enriching our understanding of MDs and mtDNA inheritance patterns. In addition, the ability of TGS to identify mtDNA chemical modifications may reveal epigenetic changes, providing insights into the impact of TGS on mitochondrial function and cell metabolism. This will further expand our understanding of the interaction between MDs, genetics, and environmental factors.
The widespread application of TGS is crucial for the task of the human pan-genome project, which aims to map all human genetic variations, including the core genome shared by different populations, the accessory genome found in specific individuals, and the unique genome unique to each person. This project transcends the limitations of single-reference genome studies, enabling us to more comprehensively capture genetic variations, especially rare variations, structural variations, and their impact on phenotypes. By comparing accessory and unique genomes, we can trace the emergence and spread of variations, elucidating how individuals adapt to environmental changes through the addition, loss, or modification of genes. This process will reveal the complex genetic structure within populations, clarify the genetic differences and evolutionary paths between populations, and provide valuable resources for disease diagnosis and personalized medicine. Cross-population and species analysis based on the pan-genome may reveal important nuclear-mitochondrial interaction mechanisms and promote a deeper exploration of the dynamics and mechanisms of endosymbiosis and NUMTs. In short, we expect that future breakthroughs in human genetic research will revolutionize medical research, disease management, and anthropological investigations. Achieving this goal requires interdisciplinary collaboration in fields such as evolutionary genomics, bioinformatics, and computer science to overcome challenges in sample bias, data processing, and analysis.
In summary, this study elaborates on the exciting research on mtDNA in recent years from the following three main aspects: the atlas of mitochondrial heteroplasmy, the distribution and impact of NUMTs, and the genetic diversity and maternal evolutionary history of mtDNA. The development of NGS, the advancement of the Human Genome Project, and the establishment of large-sample bio-databases are constantly revising and refining our understanding of mtDNA-related scientific issues. And this study proposes that in the context of the rapid development of TGS and the massive production of genomic data, multidisciplinary (especially in the field of artificial intelligence) interdisciplinary cooperation will deepen our understanding of the role of mtDNA in precision medicine, genetics, and the study of human evolutionary history, providing new perspectives and methods for solving long-standing problems in clinical medicine and anthropology.
Professor Yuan Huijun, Associate Researcher He Guanglin, and Dr. Wang Mengge from the Center for Archaeological Science, Sichuan University/West China Institute of Rare Diseases, Sichuan University, and Professor Tang Renkuan from the School of Basic Medical Sciences, Chongqing Medical University are co-corresponding authors. Master's students Luo Lintao and Liu Yunhui from Chongqing Medical University, Associate Researcher He Guanglin and Dr. Wang Mengge from the Center for Archaeological Science, Sichuan University/West China Institute of Rare Diseases, Sichuan University are co-first authors. Relevant work was supported by the National Natural Science Foundation of China, the Open Project of the Center for Archaeological Science, Sichuan University, the Major Project of the National Social Science Fund, and the 1.3.5 project of West China Hospital of Sichuan University.